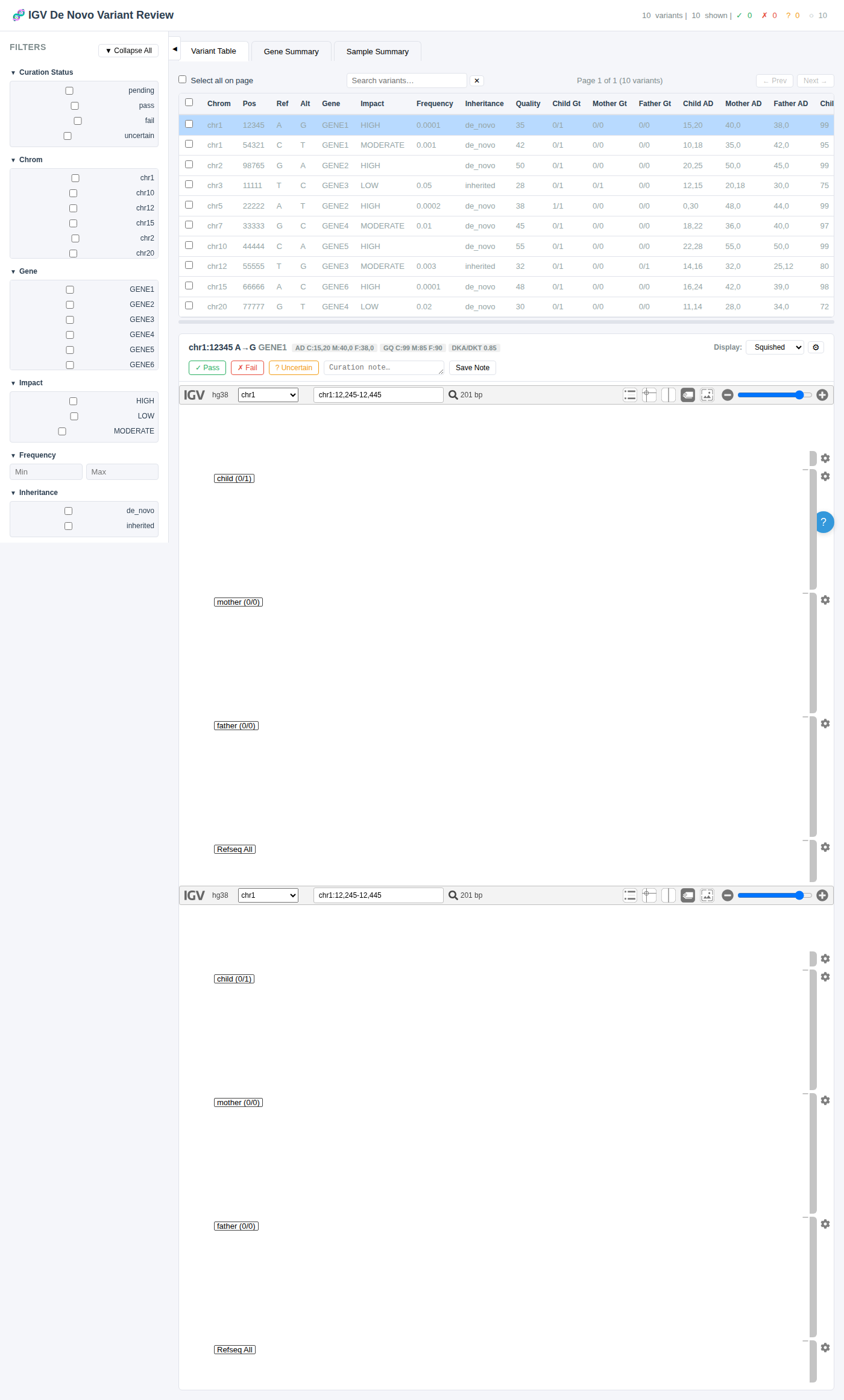
Filter Types
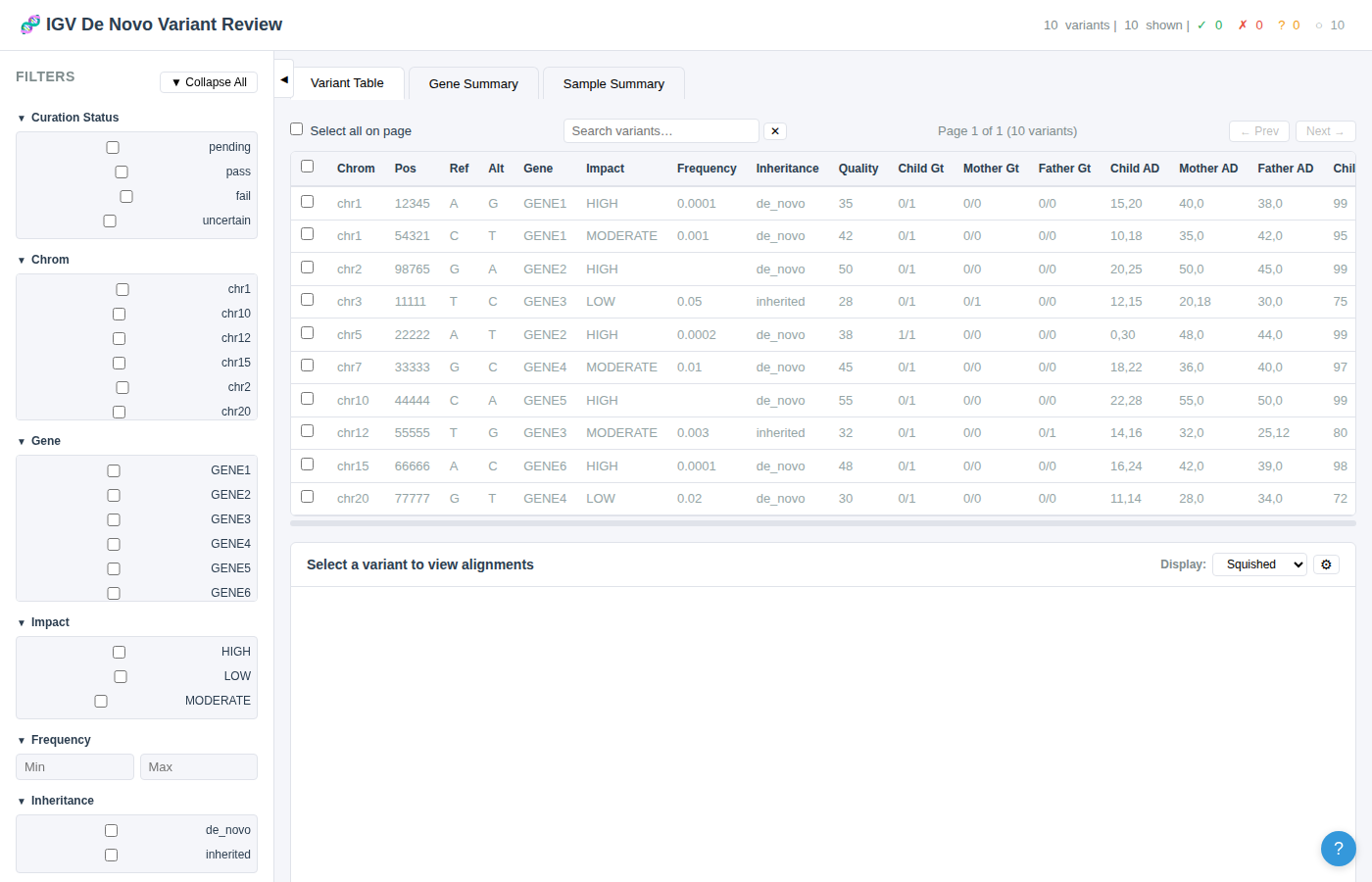
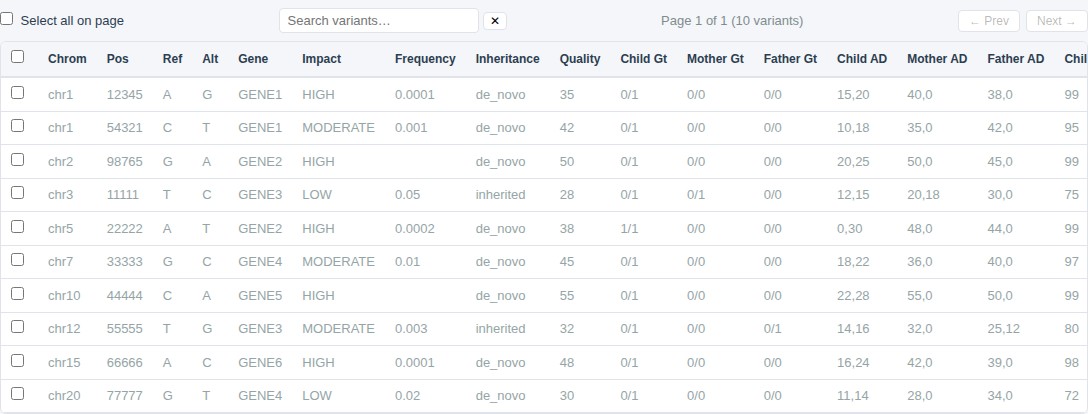
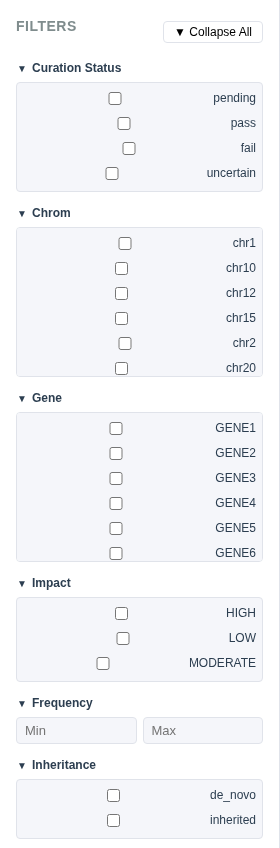
- Checkbox groups – For categorical columns with ≤10 unique values (e.g., gene, impact, inheritance, curation status). Check one or more values to include.
- Dropdown selectors – For categorical columns with >10 unique values. Select one or more from a dropdown menu.
- Range inputs – For numeric columns (e.g., frequency, quality). Set minimum and/or maximum values.
Filter Actions
- Apply Filters – Submit all current filter selections to the server and refresh the variant table.
- Clear All – Reset all filters to show all variants.
- 💾 Save Filters – Save the current filter configuration for later reuse.
- 📂 Load Filters – Restore a previously saved filter configuration.
Collapsible Groups
Each filter group can be collapsed or expanded individually by clicking the header. Use the ▼ Collapse All / ▲ Expand All button to toggle all groups at once.
Collapsible Sidebar
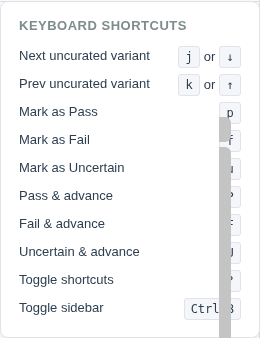
The entire filter sidebar can be collapsed using the ◀ toggle button or the keyboard shortcut Ctrl+B to maximize table and IGV viewer space.